ადამიანის გენეტიკა
მემკვიდრული დაავადებები
ნაწილი 3
ფენილკეტონურია - არის ნივთიერებათა ცვლის, კერძოდ ამინომჟავათა ფენილალანინის ცვლის მემკვიდრეობითი დარღვევა, რომელსაც თან სდევს გონებრივი ჩამორჩენილობა და სხვადასხვა ნევროლოგიური პრობლემები.
დაავადება პირველად აღწერილი იყო 1934 წელს ნორვეგიელი ექიმის ფელინგის მიერ. იგი გადაეცემა აუტოსომურ-რეცესიული გზით. ეს იმას ნიშნავს, რომ ბავშვის ორივე მშობელი დაზიანებული გენის ფარული მატარებელია. ფენილკეტონურიის მქონე ბავშვის დაბადების ალბათობა 25%-ია, ანუ ყოველი 4 მშობიარობიდან ერთი.
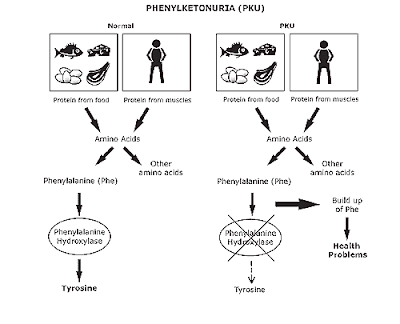
ჩვენი საკვები შედგება ცილების, ცხიმების და ნახშირწყლებისაგან. ფენილალანინი ერთ-ერთი ამინომჟავაა, რომელსაც ბავშვის ორგანიზმი ვერ უმკლავდება. ნორმალური კვებისას ზრდისა და ქსოვილებისთვის საჭირო ამინომჟავები ორგანიზმში საკმარისზე მეტია. ეს ზედმეტი ამინომჟავები სხვა ნივთიერებებად გარდაიქმნება. როგორც წესი ჭარბი ფენილალანინი გარდაიქმნება თიროზინად - სხვა ამინომჟავად, ფენილალანინ ჰიდროქსილაზას ზემოქმედებით. თიროზინის დახმარებით კი ორგანიზმში იქმნება ჰორმონები, ტვინის ქიმიური ნივთიერებები და მელანინის ყავისფერი პიგმენტი.


ფენილკეტონურიის დროს ვერ ხერხდება ფენილალანინის თიროზინად გარდაქმნა და იგი სისხლში დიდი რაოდენობით გროვდება, გარდაიქმნება ფენილკეტონებად, რომლებიც შარდში გამოიყოფა. შეიძლება მშობლებს არ ქონდეთ ფენილკეტონურია, მაგრამ დეფექტური გენის მატარებელნი იყვნენ. მათ იმიტომ არ აქვთ ფენილკეტონურია, რომ მათ ერთი დეფექტური გენი აქვთ. წყვილიდან ჯანმრთელი გენი კი საკმარისად აკონტროლებს ფენილალანინ ჰოდროქსილაზის წარმოშობას. 50-დან 1 ადამიანია ასეთი გენის მატარებელი. ორი მატარებლის შეხვედრის ალბათობა 2500-დან ერთი. შეხვედრისას ფენილკეტონურიის მქონე ბავშვის დაბადების ალბათობა ყოველ ორსულზე 25%, ფენილკეტონურიის დეფექტური გენის მქონე ბავშვის დაბადების ალბათობა 50%, ნორმალური გენის მქონე ბავშვის დაბადების ალბათობა კი 25%-ია.
ფენილკეტონურიის დროს სისხლში აღინიშნება ამინომჟავა ფენილალანინის დონის მატება - ჰიპერფენილალანინემია. მისი დონე დამოკიდებულია ფენილალანინ ჰიდროქსილაზას დეფიციტის დონეზე.
დაავადებას იწვევს ერთი ენზიმის, ფენილალანინ ჰიდროქსილაზას ნაკლებობა; ამის გამო საკვებთან ერთად მიღებული ფენილალანინი არ გადადის თიროზინში, რაც იწვევს ფენილალანინის დაგროვებას სისხლში და ზურგის ტვინის სითხეში. მისგან წარმოიქმნება ფენილ-პიტოყურძნისმჟავა და სხვა მჟავები, რომლებიც ტოქსიკურად მოქმედებენ ცენტრალურ ნეერვულ სისტემაზე.
შეერთებულ შტატებში და სხვა მრავალ ქვეყანაში ფენილკეტონურიას გამოავლენენ ახალშობილთა სკრინინგით. ამ დაავადების დროულ დიაგნოსტიკას გადამწყვეტი მნიშვნელობა აქვს ზოგადად ახალშობილის მომავალზე და ინტელექტის დონეზე. არსებობს ძალზე იშვიათი შემთხვევები როდესაც ფენილკეტონურიის დიაგნოზი პირველად ზრდასრულ ასაკში დაისვა, მაგრამ ამ დროს არ აღინიშნებოდა ინტელექტის დაქვეითება.
ფენილკეტონურიის შემთხვევათ რიცხვი იცვლება ეთნიკური ჯგუფებისა და გეოგრაფიული რეგიონების მიხედვით. შეერთებულ შტატებში ფენილკეტონურიის გავრცელების სიხშირეა 1:10000. ამ მხივ სიტუაცია თურქეთში ყველაზე რთულია, დაავადებათა სიხშირეა 4:1000. მათი უმრავლესობა გამოვლინდება დაბადებიდან მცირე ხნის შემდეგ.
დაბადებიდან პირველი სამი თვის განმავლობაში ბავშვს არაფერი ემჩნევა და რაიმეს დადგენა სპეციალური ანალიზის გარეშე რთულია. ზოგიერთ შემთხვევაში ბავშვებს დაბადებისთანავე აღენიშნებათ მაღალი აგზნებადობა (უმიზეზო ტირილი, ძილის დარღვევა), ან პირიქით, ძილიანობა, მოთენთილობა. ბავშვისათვის დამახასიათებელია უხვი ოფლიანობა და სპეციფიკური სუნი. ოთახშიც კი, სადაც ბავში იმყოფება ობის მსგავსი თავისებური სუნია.

მე-4 თვიდან შეიმჩნევა ჩამორჩენა ბავშვის ფსიქიკურ განვითარებაში: ბავშვი ხდება ინდეფერენტული მშობლების, სათამაშოების მიმართ. დროულად ვერ ჯდება, ვერ დგება, მეტყველების განვითარება ძლიერ ჩამორჩება. ბავშვების 92%- ს უვითარდება გონებრივი ჩამოჩენილობის მძიმე ხარისხი, 3-4%- ს კი უვითარდება იოლი ხარისხის გონებრივი ჩამორჩენილობა, ხოლო 0.2-0.3%-ის ფსიქიკური განვითარება ნორმის ფარგლებშია. 5-6 წლის ასაკის შემდეგ გონებრივი ჩამორჩენილობა სტაბილურ ხასიათს იღებს. ხშირად ადგილი აქვს ქცევის დარღვევას, ძლიერ აგზნებას მოტორული სტერეოტიპებით, ხანდახან ფსიქოტურ სიმპტომებსაც. ბავშვებში ერთი წლიდან ხშირია კრუნჩხვითი გულრები (50%). მათი აღმოცენება ემთხვევა ცილოვანი საკვების მიღებას. 3-5 წლისათვის კრუნჩხვითი გულყრების სიხშირე კლებულობს და ზოგ შემთხვევაში მკურნალობის გარეშეც ქრება. საშუალოდ გვხვდება 10 000 დაბადებულიდან ერთი. ერთნაირი სიხშირით გვხვდება გოგონებში და ბიჭებში. ფენილკეტონურიას ადგილი აქვს გონებრივი ჩამოჩენილობის შემთხვევების 1%-ში.
რაში მდგომარეობს ძირითადი პრობლემები ?
- მთავარი პრობლემაა ფენილალანინის ცვლის დარღვევა, მისი ჭარბი რაოდენობით დაგროვება, დიდი რაოდენობის ცილების შემვველი საკვები.
- ძირითადი პრობლემაა ბავშვის ბალანსირებული კვების დიეტა - ისეთი, რომ არ შეიცავდეს ფენილალანინს, ამავე დროს იყოს კალორიული და მოიცავდეს ბავშვის განითარებისათვის აუცილებელ ყველა ნივთიერებას.
- ბავშვის გონებრივი ჩამორჩენილობა და მასთნ დაკავშირებული ბავშვის ფსიქიკური განვითარების და სწავლის პრობლემები.
- ქცევის დარღვევა, ჰიპერაქტიულობა, გაღიზიანებადიბა, ადვილი აგზნებადობა, ხანდახან ფსიქოტური დარღვევები, იმპულსურობა.
- მეტყველების განვითარების ჩამორჩენა
- მოზრდილებში ხშირია დეპრესია
მეტად მნიშვნელოვანია ადრეული დიაგნოსტიკა. აშშ-სა და ევროპაში ახალდაბადებულ ბავშვებს მასიურად ამოწმებენ ფენილალანინზე. ყველაზე მარტივია შარდში მისი შემცველობის გასინჯვა სამქლორიანი რკინის 5%-იანი ხსნარისა და ძმარმჟავას რამდენიმე წვეთის საშუალებით. შარდი მწვანედ იღებება, თუ ფენილალანინი ნორმაზე მეტია (ფელინგის სინჯი). ფენილალანინს სისხლის პლაზმაშიც განსაზღვრავენ; მისი შემცველობა სისხლში 2-6 მგ/დლ მეტი არ უნდა იყოს. ადრეული დიაგნოსტიკა აუცილებელი პირობაა ბავშვის ნორმალური განვითარების უზრუნველყოფისთვის.
ძირითადი დახმარება მდგომარეობს ბავშვის ბალანსირებული კვების დიეტის შერჩევაში ცილოვანი საკვების მკვეთრი შემცირებით. პროტეინებით მდიდარი საკვების მიღება ან მკაცრად იზღუდება, ან საერთოდ იკრძალება. მაგრამ ფენილკეტონურიის მქონე ბავშვსაც ჭირდება გარკვეული რაოდენობის ცილები და ფენილალანინი. მისი ძლიერი დაქვეითებაც მავნეა ორგანიზმისთვის. ბავშვებს მცირე რაოდებობით აძლევენ დედის რძეს, ხილს, ბოსტნეულს, წვენებს და ფაფებს. დიეტაში შედის თითქმის ყველა ხილი, ბოსტნეულიდან კომბოსტო, სტაფილო,ჭარხალი. აგრეთვე თაფლი. რაციონიდან ამოღებულია ხორცი, თევზი, კვერცხი, ხაჭო, ყველი, ლობიო, ცერცვი, ნიგოზი. რძე და კარტოფილი კი მკვეთრად შეზღუდულია.
მუდმივი დიეტა არის ბავშვის ნორმალური გავნითარების გარანტია. მას აღარ ჩამოუალიბდება გონებრივი ჩამორჩენილობა, აღარ განუვითარდება ნევროლოგიური სიმპტომები. აღნიშნული დიეტური დახმარება ქრონიკული ხასიათისაა და მისი შეწყვეტა არ შეიძლება. წინათ ფიქრობდნენ რომ დიეტის შეწყვეტა შსესაძლებელია 10-12
წლის ასაკისათვის. მაგრამ გამოკვლევებმა აჩვენა, რომ დიეტის შეწყვეტის შემდეგ ადამიანებს ნერვული აშლილობები ეწყებათ. ამიტომ აუცილებელია ხანგრძლივი დიეტა. აუცილებელია ბავშვის სისხლის რეგულარული შემოწმება ფენილალანინის დონის განსაზღვრის მიზნით. ბავშვის ფსიქიკური განვითარების ჩამორჩენის შემთხვევაში აუცილებელია მისი სპეციალური ფსიქოლოგიური და პედაგოგიური დახმარება.
მკურნალობა ხდება დიეტის საშუალებით, რომელიც ცილების მინიმალურ რაოდენიბას შეიცავს. იმისთვის რომ ბავშვმა ზრდისთვის საჭირო რაოდენობის ფენილალანინი მიიღოს, დიეტაში შედის სპაციალური საბავშვო საკვები, რომელიც დამზადებულია ცილის კომპოზიტებზე.
ეს არის ცილის ჰიდროლიზატები. სინთეტური ფორმულა გამოიყენება აკრძალული საკვების შესაცვლელად. იგი საკმაოდ ძვირადღირებულია მაგრამ დანახარჯი შედეგს ამართლებს.
თუ დიეტა ბავშვს დროულად ენიშნება, მას არავითარი პრობლემები არ აქვს. დროული დიეტა 100%-ით იძლევა ნორმალური ფიზიკური და ფსიქიკური განვითარების გარანტიას, რადგან ფენილალანინის შემცველობა სისხლში ნორმალიზებულია. დიეტის დაცვის შემთხვევაში ფენილკეტონურიის მქონე ქალს თავისუფლად შეუძლია იყოლიოს ბავშვი. თუ მისი მეუღლე არ არის დეფექტური გენის მატარებელი, ბავშვი ჯანმრთელი დაიბადება.
ინფორმაციის წყაროები:
ლ.შანშიაშვილი. ადამიანის გენეტიკა. სალექციო მასალები.














































{kind=link}